

Comprendre les protéines, ces molécules clés dans le fonctionnement de nombreuses maladies, peut paraître simple. Il suffit d’identifier leur structure chimique et de trouver les autres protéines avec lesquelles elles peuvent se lier. Cependant, c’est là que réside toute la complexité. En effet, le domaine de recherche pour les protéines est gigantesque, nous emmenant dans un univers presque infini de possibilités de combinaisons.
« L’espace de recherche pour les protéines est énorme », a déclaré Brian Coventry, chercheur scientifique à l’Institute for Protein Design, University of Washington et The Howard Hughes Medical Institute.
Une protéine étudiée par son laboratoire est généralement composée de 65 acides aminés. Avec 20 choix différents d’acides aminés à chaque position, cela représente 65 puissance 20 combinaisons de liaison, un nombre plus grand que le nombre estimé d’atomes dans l’univers.
Une nouvelle approche innovante
Dans une étude publiée en mai 2023 dans la revue Nature Communications, Coventry et son équipe ont utilisé des méthodes d’apprentissage profond pour augmenter les modèles physiques basés sur l’énergie dans la conception computationnelle de protéines de novo. Cela a permis une augmentation de 10 fois du taux de réussite dans la liaison d’une protéine conçue avec sa protéine cible.
Leur protocole de conception de liants de protéines de novo augmenté par l’apprentissage profond comprenait des outils logiciels d’apprentissage machine tels que AlphaFold 2 et RoseTTA fold, qui a été développé par l’Institute for Protein Design.
L’importance des supercalculateurs dans la recherche
David Baker, le directeur de l’Institute for Protein Design et un chercheur du Howard Hughes Medical Institute, a bénéficié d’une allocation Pathways sur le supercalculateur Frontera du Texas Advanced Computing Center, financé par la National Science Foundation.
Le problème de l’étude était bien adapté pour la parallélisation sur Frontera, car les trajectoires de conception de protéines sont toutes indépendantes les unes des autres. Cela signifie que l’information n’avait pas besoin de passer entre les trajectoires de conception pendant que les travaux de calcul étaient en cours.
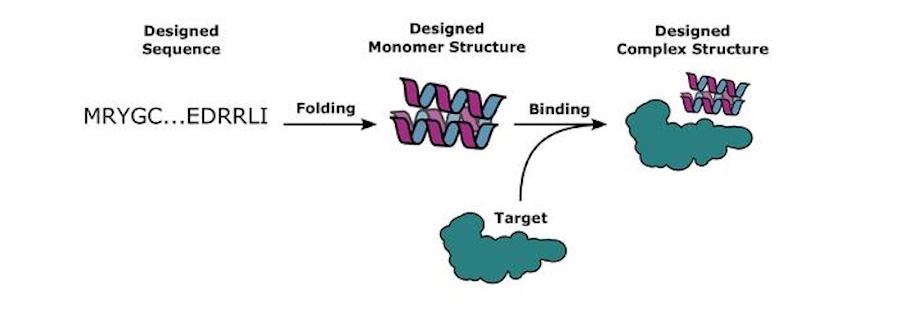
La conception de protéines à grande échelle
Les auteurs ont utilisé le programme de docking RifDock pour générer six millions de « docks » de protéines, ou interactions entre des structures de protéines potentiellement liées. Ils ont ensuite divisé ces docks en morceaux d’environ 100 000, et attribué chaque morceau à l’un des plus de 8000 nœuds de calcul de Frontera en utilisant des utilitaires Linux.
De plus, les auteurs ont utilisé l’outil logiciel Protein MPNN développé par l’Institute for Protein Design pour augmenter encore l’efficacité de la génération de séquences de protéines avec des réseaux neuronaux à plus de 200 fois plus vite que le meilleur logiciel précédent.
En synthèse
Même si les résultats de l’étude ont montré une augmentation de 10 fois du taux de réussite pour les structures conçues pour se lier à leur protéine cible, le chemin est encore long, selon Coventry. L’avenir de la recherche est d’augmenter encore ce taux de réussite et de passer à une nouvelle classe de cibles encore plus difficiles. Les virus et les récepteurs des cellules T du cancer sont des exemples parfaits.
Pour une meilleure compréhension
Q: Qu’est-ce que l’apprentissage profond en conception de protéines ?
C’est une méthode qui utilise des algorithmes informatiques pour analyser et tirer des inférences à partir de motifs dans les données. Dans cette étude, ces méthodes ont été utilisées pour apprendre des transformations itératives de représentation de la séquence de protéines et de la structure possible.
Q: Quels sont les avantages de l’utilisation de supercalculateurs dans cette recherche ?
Les supercalculateurs permettent de traiter un grand nombre de trajectoires de conception en parallèle, ce qui permet d’accélérer significativement le processus de recherche.
Q: Quel est l’objectif final de cette recherche ?
L’objectif est d’augmenter le taux de réussite de la conception de protéines et de développer des médicaments plus efficaces contre des maladies comme le cancer et les virus.
L’étude, intitulée « Improving de novo protein binder design with deep learning », a été publiée le 6 mai 2023 dans la revue Nature Communications. Les coauteurs sont Nathaniel R. Bennett, Brian Coventry, Inna Goreshnik, Buwei Huang, Aza Allen, Dionne Vafeados, Ying Po Peng, Justas Dauparas, Minkyung Baek, Lance Stewart, Frank DiMaio et David Baker de l’université de Washington ; Steven De Munck et Savvas N. Savvides de l’université de Gand. DOI : 10.1038/s41467-023-38328-5













